

500 mg
For the use of a Registered Medical Practitioner or a Hospital or a Institution only.
SODIUM FUSIDATE FOR INJECTION (Sodium Fusidate) is an antimicrobial substance produced by the growth of certain strains of Fusidium coccineum. Chemically, Sodium Fusidate is Sodium (Z)-ent-16a-(acetyloxy)-3b,11b-dihydroxy-4b, 8, 14-trimethyl-18-nor-5b,10a-cholesta-17(20), 24-dien-21-oate. The molecular formula is C31H47NaO6 and molecular weight is 538.7.
STRUCTURAL FORMULA :
Its structural formula is :
.jpg)
SODIUM FUSIDATE FOR INJECTION is sterile, white to almost white crystalline powder filled in vial of suitable size.
COMPOSITION :
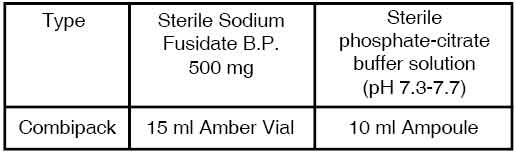
This Combipack contains :
a) One vial of
Sterile Sodium Fusidate B.P. 500 mg
b) One Ampoule of 10 ml
Sterile Phosphate-Citrate Buffer Solution
Each ampoule contains :
Disodium Phosphate (Dihydrate)
Citric Acid (Monohydrate)
Disodium Edetate
Water for Injection
pH adjusted between 7.3 to 7.7
ACTIONS :
Fusidic acid is a potent antibiotic derived from the fungus Fusidium coccineum. Its mode of action is by inhibition of protein synthesis by the prevention of translocation on the ribosome. Concentrations adequate for bactericidal activity against staphylococci have been demonstrated in the following : pus, exudate, soft tissue, bone tissue, synovial fluid, aqueous humour, vitreous body, burn crusts, intracranial abscess, sputum, serum. Fusidic acid is structurally related to cephalosporin P and helvolic acid, neither of which has been developed for clinical use. SODIUM FUSIDATE FOR INJECTION exerts antibacterial activity against most Gram-positive organisms; in particular, it is effective against pathogenic staphylococci, including penicillinase producing and methicillin-resistant strains. The MICs for most Staph aureus strains are between 0.02-0.12 mg/l. It is much less active against S. pyogenes with the MIC between 4-20 mg/l. It has slight or no activity against Gram-negative organisms and fungi.
Both oral and intravenous Sodium Fusidate have been given in combination with other antibiotics, e.g. cloxacillin, cephaloridine, ampicillin, methicillin, erythromycin, novobiocin, rifampicin. Such a combination may prevent the development of Sodium Fusidate resistant strains. No cross-resistance occurs between Sodium Fusidate and any other antibiotic in clinical use. Because it is predominantly effective against Gram-positive organisms, disturbance of the normal gastrointestinal flora is unlikely.
PHARMACOKINETICS :
500 mg of sodium fusidate given as a single infusion over 2 hours results in a Cmax of 52 micrograms/ml. Blood levels are cumulative, reaching concentrations of 60-120 micrograms/ml after repeated infusion of 500 mg sodium fusidate every 8 hours for 2-3 days. The plasma half-life is approximately 10-15 hours. Fusidic acid is extensively excreted in the bile; 0.3 % is present as the unmetabolized drug, 15 % is the glucuronide conjugate, 10 % is the dicarboxylic metabolite and 3 % is the hydroxy metabolite. Fusidic acid is excreted 20 % in the faeces.
INDICATIONS :
This product is indicated in the treatment of all staphylococcal infections due to susceptible organisms such as :Osteomyelitis, pneumonia, septicaemia, wound infections, endocarditis, superinfected cystic fibrosis, cutaneous infections. It should be administered intravenously whenever oral therapy is inappropriate, which includes cases where absorption from the gastro-intestinal tract is unpredictable. If bacteriological diagnosis reveals methicillin-resistant Staphylococcus aureus, the use of SODIUM FUSIDATE FOR INJECTION monotherapy is not appropriate and concurrent treatment with other anti-staphylococcal antibiotics is necessary.
Administration :
FOR INTRAVENOUS INFUSION AFTER DILUTION.
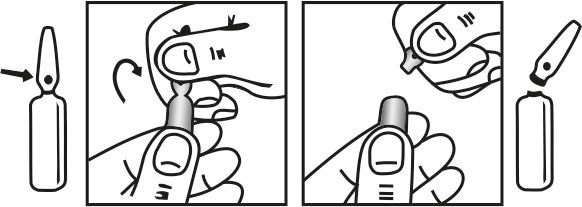
INSTRUCTIONS FOR USE OF AMPOULE :
The ampoule used in this product is equipped with O.P.C (One Point Cut) opening system. No ampoule file is needed to open the ampoule. The neck of the ampoule is prescored at the point of constriction. A coloured dot on the ampoule head helps to orientate the ampoule. Take the ampoule and face the coloured dot. Let the solution at the head of the ampoule to flow down by shaking or a gentle stroke. The ampoule opens easily by placing the thumb on the coloured dot and gently pressing downwards as shown.
Dosage :
Adults weighing more than 50 kg : 500 mg sodium fusidate three times daily. Children and adults weighing less than 50 kg : 6-7 mg sodium fusidate per kg bodyweight three times daily.
Recommended procedure : To reconstitute, dissolve the contents of one vial containing 500 mg sodium fusidate powder (equivalent to 480 mg of fusidic acid) in the 10 ml buffer provided.
For adults weighing more than 50 kg : Add the 10 ml fusidate/buffer solution to 500 ml of infusion fluid.
For children and adults weighing less than 50 kg : Add the 10 ml fusidate/buffer solution to 500 ml of infusion fluid. Each dose corresponds to 6-7 ml of the resulting solution per kg bodyweight.The diluted fluid should be infused via a central venous line over 2 hours. If a superficial vein is employed a more prolonged period of at least 6 hours is advisable. This product should be administered intravenously into a wide bore vein with a good blood flow. Excessive doses may cause venospasm, thrombophlebitis and haemolysis of erythrocytes. Both oral and intravenous presentations have been given concurrently with other antibiotics, e.g. cloxacillin, flucloxacillin, ampicillin, methicillin and erythromycin. Since it is excreted in the bile, no dosage modifications are needed in renal impairment. The dosage in patients undergoing haemodialysis needs no adjustment as this product is not significantly dialysed.
Dosage in the elderly : No dosage alterations are necessary in the elderly. If additional antibacterial therapy is to be employed, it is recommended that for parenteral administration, separate infusion fluids be used.
CONTRAINDICATIONS :
Hypersensitivity to fusidic acid and its salts, or any of the excipients. This product should not be infused with amino acid solutions or in whole blood. Due to local tissue injury, this product should not be administered intramuscularly or subcutaneously.
WARNINGS AND PRECAUTIONS :
SODIUM FUSIDATE FOR INJECTION must not be administered intramuscularly (IM) or subcutaneously because of high incidence of local tissue injury following IM route. SODIUM FUSIDATE FOR INJECTION should only be used for IV infusion SODIUM FUSIDATE FOR INJECTION must not be co-administered with statins. There have been reports of rhabdomyolysis (including some fatalities) in patients receiving this combination. In patients where the use of systemic SODIUM FUSIDATE FOR INJECTION is considered essential, statin treatment should be discontinued throughout the duration of SODIUM FUSIDATE FOR INJECTION treatment. The patient should be advised to seek medical advice immediately if they experience any symptoms of muscle weakness, pain or tenderness. Statin therapy may be re-introduced seven days after the last dose of SODIUM FUSIDATE FOR INJECTION. In exceptional circumstances, where prolonged systemic SODIUM FUSIDATE FOR INJECTION is needed e.g. for the treatment of severe infections, the need for co-administration of statin and SODIUM FUSIDATE FOR INJECTION should only be considered on a case by case basis and under close medical supervision. Sodium fusidate is metabolised in the liver and excreted in the bile. Caution should be exercised with other antibiotics which have similar biliary excretion pathways e.g. lincomycin and rifampicin. Elevated liver enzymes and jaundice have occurred during systemic therapy but are usually reversible on discontinuation of the drug.
Periodic liver function tests should be carried out when the product is given :
• in high oral doses
• for prolonged periods
• to patients with liver dysfunction
• to patients taking potentially hepatotoxic medication
• to patients with biliary tract obstruction
• to patients taking concurrent medication with a similar excretion pathway.
Sodium fusidate displaces bilirubin from its albumin binding site in vitro. Caution is necessary if this product is administered to patients with impaired transport and metabolism of bilirubin. The use of this product in combination with drugs that are CYP-3A4 biotransformed should be avoided. Bacterial resistance has been reported to occur with the use of sodium fusidate. As with all antibiotics, extended or recurrent use may increase the risk of developing antibiotic resistance. This medicinal product contains 3.2 mmol (73 mg) sodium per 500 mg dose. This should be taken into consideration by patients on a controlled sodium diet.
Pregnancy : Category C
Fusidic acid may cause kernicterus in babies during the first month of life by displacing bilirubin from plasma albumin. Fusidic acid should be avoided when possible during the last month of pregnancy.
Nursing mothers :
Safety in lactation has not been established. There is evidence that the drug can penetrate the placental barrier and is detectable in human milk. Caution is therefore required when SODIUM FUSIDATE FOR INJECTION is used in mothers who wish to breast feed.
Paediatric Use :
SODIUM FUSIDATE FOR INJECTION has been given to neonates from the day of birth and for periods of up to one year without adverse effects. Despite the immature enzyme system in the neonate, none of the children showed any evidence of hepatotoxicity, nor did they develop renal, blood, ocular or other toxicity.
INTERACTIONS AND INCOMPATIBILITIES :
Interactions :
The risk of myopathy including rhabdomyolysis may be increased by the concomitant administration of systemic SODIUM FUSIDATE FOR INJECTION with statins. Co administration of this combination may cause increased plasma concentrations of both agents. The mechanism of this interaction (whether it is pharmacodynamics or pharmacokinetic, or both) is yet unknown. There have been reports of rhabdomyolysis (including some fatalities) in patients receiving this combination. If treatment with SODIUM FUSIDATE FOR INJECTION is necessary, statin treatment should be discontinued throughout the duration of the SODIUM FUSIDATE FOR INJECTION treatment. In vitro compatibility studies of Sodium Fusidate 500 mg for Intravenous Infusion with commonly used infusion solutions have been carried out. The results showed that sodium fusidate reconstituted at 50 mg/ml in buffer solution is physically and chemically compatible for at least 24 hours at room temperature with the following infusion solutions (the figure in parenthesis shows the concentration of sodium fusidate in the final admixture) :
Sodium Chloride Intravenous Infusion B.P. 0.9 % (1-2 mg/ml). Dextrose Intravenous Infusion B.P. 5 % (1-2 mg/ml). Compound Sodium Lactate Intravenous Infusion (“Ringer-Lactate Solution”) (1 mg/ml). Sodium Lactate Intravenous Infusion B.P. (1 mg/ml). Sodium Chloride (0.18 %) and Dextrose (4 %) Intravenous Infusion B.P. (1 mg/ml). Potassium Chloride (0.3 %) and Dextrose (5 %) Intravenous Infusion B.P. (1 mg/ml). Specific pathways of metabolism of this product in the liver are not known, however, an interaction between this product and drugs being CYP-3A4 biotransformed can be suspected. The mechanism of this interaction is presumed to be a mutual inhibition of metabolism. There is insufficient data to characterise the effect of fusidic acid on CYPs in-vitro. The use of this product systemically should be avoided in patients treated with CYP-3A4 biotransformed drugs.
When this product is administered systemically and concomitantly with oral anticoagulants such as coumarin derivatives or anticoagulants with similar actions, the plasma concentration of these agents may increase enhancing the anticoagulant effect. Anticoagulation should be closely monitored and a decrease of the oral anticoagulant dose may be necessary in order to maintain the desired level of anticoagulation. Similarly, discontinuation of this product may require the maintenance dose of anticoagulant to be re-assessed. The mechanism of this suspected interaction remains unknown. Co-administration of this product and HIV protease inhibitors such as ritonavir and saquinavir causes increased plasma concentrations of both agents which may result in hepatotoxicity. Co-administration of this product systemically with ciclosporin has been reported to cause increased plasma concentration of ciclosporin.
Incompatibilities :
Sodium fusidate reconstituted at 50 mg/ml in buffer solution is physically incompatible with infusion fluids containing 20 % or more of dextrose, lipid infusions and peritoneal dialysis fluids. Precipitation may occur at dilutions which result in a pH of less than 7.4.
SIDE EFFECTS :
Based on clinical trial data on sodium fusidate administered intravenously, in high doses and concomitantly with other antibiotics in critically ill patients, it is estimated that approximately 30 % of the patients may experience an undesirable effect. This number is reduced when the product is administered through a central vein. Venous intolerance such as venous spasm and thrombophlebitis are very common when the product is administered through a peripheral vein, while common when it is administered through a central line. Raised bilirubin, liver enzymes and clinical jaundice are considered to be common. These undesirable effects are usually reversible on discontinuation of the drug. Side effects are listed below, by MedDRA System Organ Class, in decreasing order of frequency within each class. Where frequencies are given, these are based on the clinical trial data, using the stated frequency classification. Where the term ‘Not known’ is given, these effects are derived from spontaneous reports.
OVERDOSAGE :
The following effects have been selected on the basis of their potential clinical significance (possible signs and symptoms in parentheses where appropriate)-not necessarily inclusive :
Acute :
Diarrhoea or (increase in bowel movements; loose stools; soft stools), epigastric or gastric discomfort.
Chronic :
Abnormal liver biochemistry Jaundice (itching; loss of appetite; nausea; abdominal or stomach pain; unusual tiredness or weakness; yellow eyes or skin)
TREATMENT OF OVERDOSAGE :
Monitoring :
Excessive intravenous doses of fusidic acid may result in hypocalcaemia due to the amount of phosphate buffer administered (each 10 millilitres of phosphate/citrate buffer for intravenous infusion contains 196 milligrams disodium hydrogen phosphate equivalent to 1.1 millimols of phosphate) Patient monitoring including serum calcium determinations and possible treatments may be required with intravenous overdoses of fusidic acid. Patients in whom intentional overdose is confirmed or suspected should be referred for psychiatric consultation.
PHARMACEUTICAL PRECAUTIONS :
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
STORAGE :
Store below 25° C (77°F), protected from moisture and light.
Do not refrigerate.
The sodium fusidate dry powder is stable for 2 years when stored at room temperature (below 25°C) and protected from light. When the buffer solution is transferred to the powder vial, this vial should be regarded as a unit dose. The required amount of the sodium fusidate/buffer solution should be used once only and any unused portion discarded.
SHELF LIFE :
24 months from the date of manufacture.
PRESENTATION :
SODIUM FUSIDATE FOR INJECTION is supplied as :
Disclaimer : For the use of a Registered Medical Practitioner or a Hospital or a Institution only. Also it is not intended to be used by healthcare professionals or patients for the purpose of prescribing or administering these products. Questions regarding the complete and current content of product labeling / specification / presentation should be directed to SGPharma.