


50 mg/2 ml
For the use of a Registered Medical Practitioner or a Hospital or a Institution only.
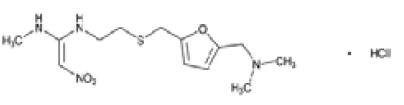
RANITIDINE INJECTION USP (Ranitidine Hydrochloride) is a histamine H2-receptor antagonist. Chemically, Ranitidine Hydrochloride is N-[2-[[[5-[(Dimethylamino)methyl]-2-furanyl]methyl]thio]ethyl]-N’-methyl-2-nitro-1,1-ethenediamine, hydrochloride. The molecular formula is C13H22N4O3S·HCl and molecular weight is 350.86.
STRUCTURAL FORMULA :
Its structural formula is :
RANITIDINE INJECTION USP is a sterile, clear, colourless to yellow, nonpyrogenic solution filled in amber ampoule of suitable size.
COMPOSITION :
Each ml contains :
Ranitidine Hydrochloride USP
equivalent to Ranitidine 25 mg
Phenol USP 0.5 % w/v
(as preservative)
Water for Injection USP q.s.
Dibasic Sodium Phosphate USP and Monobasic Potassium Phosphate USP are added as buffers.
ACTIONS :
Ranitidine is a specific, rapidly acting histamine H2-antagonist. It inhibits basal and stimulated secretion of gastric acid, reducing both the volume and the acid and pepsin content of the secretion.
PHARMACOKINETICS :
Ranitidine is rapidly absorbed on intramuscular injection, with peak plasma concentrations occurring in about 15 minutes. It is weakly bound, about 15 %, to plasma proteins. The elimination half-life is about 2 to 3 hours and is increased in renal impairment. A small proportion of ranitidine is metabolised in the liver to the N-oxide, the S-oxide, and desmethylranitidine; the N-oxide is the major metabolite but accounts for only about 4 to 6 % of a dose. About 30 % of an oral dose and 70 % of an intravenous dose is excreted unchanged in the urine in 24 hours, primarily by active tubular secretion; there is some excretion in the faeces. Ranitidine crosses the placental barrier and is distributed into breast milk.
Special Patient Populations :
Children/infants (6 months and above) :
Limited pharmacokinetic data show that there were no significant differences in half-life (range for children 3 years and above : 1.7 - 2.2 h) and plasma clearance (range for children 3 years and above : 9-22 ml/min/kg) between children and healthy adults receiving intravenous ranitidine when correction is made for body weight. Pharmacokinetic data in infants is extremely limited but appears to be in line with that for older children.
Patients over 50 years of age :
In patients over 50 years of age, half-life is prolonged (3-4 h) and clearance is reduced, consistent with the age-related decline of renal function. However, systemic exposure and accumulation are 50 % higher. This difference exceeds the effect of declining renal function, and indicates increased bioavailability in older patients.
Neonates (under 1 month) :
Limited pharmacokinetic data from term babies undergoing treatment with Extracorporeal Membrane Oxygenation (ECMO) suggests that plasma clearance following intravenous administration may be reduced (1.5-8.2 ml/min/kg) and the half-life increased in the new-born. Clearance of ranitidine appeared to be related to the estimated glomerular filtration rate in the neonates.
INDICATIONS :
RANITIDINE INJECTION USP is indicated for the treatment of duodenal ulcer, benign gastric ulcer, post-operative ulcer and of Zollinger - Ellison Syndrome. In the management of conditions where reduction of gastric secretion and acid output is desirable, such as reflux oesophagitis.
As prophylaxis against :
gastrointestinal haemorrhage from stress ulceration in seriously ill patients. recurrent haemorrhage in patients with bleeding peptic ulcers. acid aspiration (Mendelson’s Syndrome) before anaesthesia in patients at risk, particularly obstetric patients during labour.
Children (6 months to 18 years) :
Short term treatment of peptic ulcer. treatment of gastro-oesophageal reflux, including reflux oesophagitis and symptomatic relief of gastro-oesophageal reflux disease.
Administration :
RANITIDINE INJECTION USP is given by intramuscular injection or slow intravenous injection or intravenous infusion.
RANITIDINE INJECTION USP has been shown to be compatible with the following intravenous infusion fluids :
- Sodium Chloride 0.9 % w/v
- Dextrose 5 % w/v
- Sodium Chloride 0.18 % w/v and Dextrose 4 % w/v
- Sodium Bicarbonate 4.2 % w/v
- Hartmann’s solution
From a microbiological point of view, the product should be used immediately. If not used immediately, in-use storage times and conditions prior to use are the responsibility of the user and would normally not be longer than 24 hours at 2 to 8°C, unless preparation of solutions has taken place in controlled and validated aseptic conditions. All solutions of RANITIDINE INJECTION USP should be discarded after use.
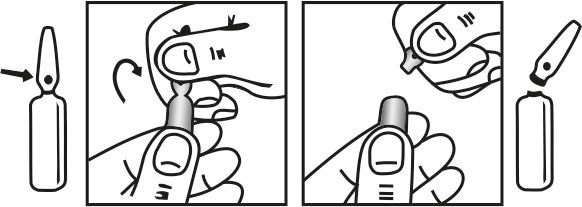
INSTRUCTIONS FOR USE OF AMPOULE :
The ampoule used in this product is equipped with O.P.C. (One Point Cut) opening system. No ampoule file is needed to open the ampoule. The neck of the ampoule is prescored at the point of constriction. A coloured dot on the ampoule head helps to orientate the ampoule. Take the ampoule and face the coloured dot. Let the solution at the head of the ampoule to flow down by shaking or a gentle stroke. The ampoule opens easily by placing the thumb on the coloured dot and gently pressing downwards as shown.
Dosage :
Adults (including elderly) and adolescents (12 years and older) :
RANITIDINE INJECTION USP Solution for Injection may be given as :
a slow intravenous injection (over two minutes) up to a maximum of 50 mg, after dilution to a volume of 20 ml per 50 mg dose. This dose may be repeated every 6 to 8 hours; or as an intermittent intravenous infusion at a rate of 25 mg per hour for two hours. The infusion may be repeated at 6 to 8 hour intervals; or as an intramuscular injection of 50 mg (2 ml) every 6 to 8 hours.
Prophylaxis of haemorrhage from stress ulceration or recurrent haemorrhage :
In the prophylaxis of haemorrhage from stress ulceration in seriously ill patients or the prophylaxis of recurrent haemorrhage in patients bleeding from peptic ulceration, parenteral administration may be continued until oral feeding commences. Patients considered to be still at risk may then be treated orally with Ranitidine tablets 150 mg twice daily. In the prophylaxis of upper gastro-intestinal haemorrhage from stress ulceration in seriously ill patients a priming dose of 50 mg as a slow intravenous injection followed by a continuous intravenous infusion of 0.125 - 0.250 mg/kg/hr may be preferred.
Prophylaxis of Mendleson’s syndrome :
In patients considered at risk of developing acid aspiration (Mendelson’s) syndrome, RANITIDINE INJECTION USP may be given intramuscularly or by slow intravenous injection, 45 to 60 minutes before induction of general anaesthesia.
Children / Infant (6 months to 11 years) :
RANITIDINE INJECTION USP may be given as a slow (over 2 minutes) intravenous injection up to a maximum of 50 mg every 6 to 8 hours.
Peptic Ulcer Acute Treatment and Gastro-Oesophageal Reflux :
Intravenous therapy in children with peptic ulcer disease is indicated only when oral therapy is not possible. For acute treatment of peptic ulcer disease and gastro-oesophageal reflux in paediatric patients, RANITIDINE INJECTION USP may be administered at doses that have been shown to be effective for these diseases in adults and effective for acid suppression in critically ill children. The initial dose (2.0 mg/kg or 2.5 mg/kg, maximum 50 mg) may be administered as a slow intravenous infusion over 10 minutes, either with a syringe pump followed by a 3 ml flush with normal saline over 5 min, or following dilution with normal saline to 20 ml. Maintenance of pH > 4.0 can be achieved by intermittent infusion of 1.5 mg/kg every 6 h to 8 h. Alternatively treatment can be continuous, administering a loading dose of 0.45 mg/kg followed by a continuous infusion of 0.15 mg/kg/hr.
Renal Impairment :
Accumulation of ranitidine with resulting elevated plasma concentrations will occur in patients with renal impairment (creatinine clearance less than 50 ml/min). It is recommended in such patients that ranitidine be administered in doses of 25 mg.
CONTRAINDICATIONS :
RANITIDINE INJECTION USP is contraindicated for patients known to have hypersensitivity to the active substance or any of the excipients.
Treatment with a histamine H2-antagonist may mask the symptoms associated with carcinoma of the stomach and may therefore delay diagnosis of the condition. Where gastric ulcer is suspected, the possibility of malignancy should be excluded before therapy with ranitidine is started. Ranitidine is excreted via the kidney and so plasma levels of the drug are increased in patients with renal impairment. Bradycardia in association with rapid administration of RANITIDINE INJECTION USP has been reported rarely, usually in patients with factors predisposing to cardiac rhythm disturbances. Recommended rates of administration should not be exceeded. It has been reported that the use of higher than recommended doses of intravenous H2-antagonists has been associated with rises in liver enzymes when treatment has been extended beyond five days.
Although clinical reports of acute intermittent porphyria associated with ranitidine administration have been rare and inconclusive, ranitidine should be avoided in patients with a history of this condition. In patients such as the elderly, persons with chronic lung disease, diabetes or the immunocompromised, there may be an increased risk of developing community acquired pneumonia. A large epidemiological study showed an increased risk of developing community acquired pneumonia in current users of H2 receptor antagonists versus those who had stopped treatment, with an observed adjusted relative risk increase of 1.82 (95 % CI, 1.26–2.64). Post marketing data indicate reversible mental confusion, depression, and hallucinations have been reported most frequently in severely ill and elderly patients.
Laboratory Test :
False-positive tests for urine protein with Urine Reagent Test Strips may occur during therapy with ranitidine, and therefore testing with sulfosalicylic acid is recommended.
Pregnancy : Category B
Ranitidine crosses the placenta. Adequate and well-controlled studies in humans have not been done. Studies in rats and rabbits at doses up to 160 times the human dose have not shown that ranitidine causes adverse effects on the foetus.
Nursing mothers :
Ranitidine is secreted in human milk. Caution should be exercised when RANITIDINE INJECTION USP is administered to a nursing mother.
Paediatric use :
The safety and effectiveness of RANITIDINE INJECTION USP have been established in the age-group of 1 month to 16 years for the treatment of duodenal ulcer. Use of RANITIDINE INJECTION USP in this age-group is supported by adequate and well-controlled studies in adults, as well as additional pharmacokinetic data in paediatric patients, and an analysis of the published literature. Safety and effectiveness in paediatric patients for the treatment of pathological hypersecretory conditions have not been established. Limited data in neonatal patients (less than 1 month of age) receiving ECMO suggest that ranitidine hydrochloride may be useful and safe for increasing gastric pH for patients at risk of gastrointestinal haemorrhage.
INTERACTIONS AND INCOMPATIBILITIES :
Ranitidine has the potential to affect the absorption, metabolism or renal excretion of other drugs. The altered pharmacokinetics may necessitate dosage adjustment of the affected drug or discontinuation of treatment.
Interactions occur by several mechanisms including :
1) Inhibition of cytochrome P450-linked mixed function oxygenase system :
Ranitidine, at blood levels produced by standard doses, does not inhibit or interact significantly with the hepatic cytochrome P450-linked mixed function oxygenase system. Accordingly, ranitidine in usual therapeutic doses, does not potentiate the actions of drugs which are inactivated by this enzyme; these include diazepam, lidocaine, phenytoin, propranolol and theophylline. There have been reports of altered prothrombin time with coumarin anticoagulants (e.g. warfarin). Due to the narrow therapeutic index, close monitoring of increased or decreased prothrombin time is recommended during concurrent treatment with ranitidine.
Competition for renal tubular secretion :
Since ranitidine is partially eliminated by the cationic system, it may affect the clearance of other drugs eliminated by this route. High doses of ranitidine (e.g. such as those used in the treatment of Zollinger-Ellison syndrome) may reduce the excretion of procainamide and N-acetylprocainamide resulting in increased plasma level of these drugs.
3) Alteration of gastric pH :
The bioavailability of certain drugs may be affected. This can result in either an increase in absorption (e.g. triazolam, midazolam, glipizide) or a decrease in absorption (e.g. ketoconazole, atazanavir, delavirdine, gefitinib).
SIDE EFFECTS :
Transient pain at the site of intramuscular injection has been reported. Transient local burning or itching has been reported with intravenous administration of RANITIDINE INJECTION USP. The following have been reported as events in clinical trials or in the routine management of patients treated with oral or parenteral RANITIDINE INJECTION USP. The relationship to therapy with RANITIDINE INJECTION USP has been unclear in many cases. Headache, sometimes severe, seems to be related to administration of RANITIDINE INJECTION USP.
Central Nervous System :
Rarely, malaise, dizziness, somnolence, insomnia, and vertigo. Rare cases of reversible mental confusion, agitation, depression, and hallucinations have been reported, predominantly in severely ill elderly patients. Rare cases of reversible blurred vision suggestive of a change in accommodation have been reported. Rare reports of reversible involuntary motor disturbances have been received.
Cardiovascular :
As with other H2-blockers, rare reports of arrhythmias such as tachycardia, bradycardia, asystole, atrioventricular block, and premature ventricular beats.
Gastrointestinal :
Constipation, diarrhoea, nausea/vomiting, abdominal discomfort/pain, and rare reports of pancreatitis.
Hepatic :
In normal volunteers, SGPT values were increased to at least twice the pretreatment levels in 6 of 12 subjects receiving 100 mg intravenously 4 times daily for 7 days, and in 4 of 24 subjects receiving 50 mg intravenously 4 times daily for 5 days. There have been occasional reports of hepatocellular, cholestatic, or mixed hepatitis, with or without jaundice. In such circumstances, ranitidine should be immediately discontinued. These events are usually reversible, but in rare circumstances death has occurred. Rare cases of hepatic failure have also been reported.
Musculoskeletal :
Rare reports of arthralgias and myalgias.
Haematologic :
Blood count changes (leucopenia, granulocytopenia, and thrombocytopenia) have occurred in a few patients. These were usually reversible. Rare cases of agranulocytosis, pancytopenia, sometimes with marrow hypoplasia, and aplastic anaemia and exceedingly rare cases of acquired immune haemolytic anaemia have been reported.
Endocrine :
Controlled studies in animals and humans have shown no stimulation of any pituitary hormone by RANITIDINE INJECTION USP and no antiandrogenic activity, and cimetidine-induced gynaecomastia and impotence in hypersecretory patients have resolved when RANITIDINE INJECTION USP has been substituted. However, occasional cases of impotence and loss of libido have been reported in male patients receiving RANITIDINE INJECTION USP, but the incidence did not differ from that in the general population. Rare cases of breast symptoms and conditions, including galactorrhoea and gynaecomastia, have been reported in both males and females.
Integumentary :
Rash, including rare cases of erythema multiforme. Rare cases of alopecia and vasculitis.
Respiratory :
A large epidemiological study suggested an increased risk of developing pneumonia in current users of histamine-2-receptor antagonists (H2RAs) compared to patients who had stopped H2RA treatment, with an observed adjusted relative risk of 1.63 (95 % CI, 1.07 to 2.48). However, a causal relationship between use of H2RAs and pneumonia has not been established.
Other :
Rare cases of hypersensitivity reactions (e.g., bronchospasm, fever, rash, eosinophilia), anaphylaxis, angioneurotic oedema, acute interstitial nephritis, and small increases in serum creatinine.
EFFECTS ON ABILITY TO DRIVE AND USE MACHINES :
None known.
OVERDOSAGE :
Ranitidine is very specific in action and no particular problems are expected following overdosage with the medicine.
TREATMENT OF OVERDOSAGE :
There is no specific antidote for overdose with histamine H2-receptor antagonists. Symptomatic and supportive therapy should be given as appropriate.
PHARMACEUTICAL PRECAUTIONS :
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
STORAGE :
Store below 30°C (86°F), protected from light.
Do not freeze.
RANITIDINE INJECTION USP tends to exhibit a yellow colour that may intensify over time without adversely affecting potency.
SHELF LIFE :
24 months from the date of manufacture.
PRESENTATION :
RANITIDINE INJECTION USP is supplied as Ranitidine Hydrochloride USP equivalent to Ranitidine 50 mg in 2 ml aqueous solution.
Such 5 ampoules of 2 ml are packed in a Box.
Disclaimer : For the use of a Registered Medical Practitioner or a Hospital or a Institution only. Also it is not intended to be used by healthcare professionals or patients for the purpose of prescribing or administering these products. Questions regarding the complete and current content of product labeling / specification / presentation should be directed to SGPharma.



