

250 mg, 500 mg
For the use of a Registered Medical Practitioner or a Hospital or a Institution only.
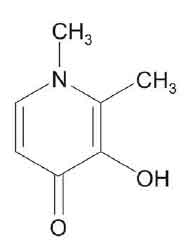
DEFERIPRONE CAPSULES (Deferiprone) is an orally active iron chelator used in the treatment of iron overload in patients with thalassaemia for whom desferrioxamine is unsuitable or ineffective. Chemically, Deferiprone is 3-Hydroxy-1,2-dimethyl-4(1H)-pyridinone. The molecular formula is C7H9NO2 and molecular weight is 139.15.
STRUCTURAL FORMULA :
Its structural formula is :
DEFERIPRONE CAPSULES 250 mg contains almost white coloured powder filled in hard gelatin capsule of suitable size. DEFERIPRONE CAPSULES 500 mg contains almost white coloured powder filled in hard gelatin capsule of suitable size.
COMPOSITION :
Each hard gelatin capsule contains :
Deferiprone 250 mg
Excipients q.s.
Approved colours used in empty capsule shells
Each hard gelatin capsule contains :
Deferiprone 500 mg
Excipients q.s.
Approved colours used in empty capsule shells
ACTIONS :
Deferiprone (L1) is an oral iron chelating agent belonging to the group of hydroxypyridones. Deferiprone forms neutral complexes with iron at physiological pH. It mobilizes iron from the iron storage proteins ferritin and haemosiderin; and from iron saturated transferrin and lactoferrin, but not from haemoglobin and myoglobin. The water soluble complex of iron formed is rapidly excreted in the urine thus reducing pathological iron deposits in organs and tissues.
PHARMACOKINETICS :
Absorption :
Deferiprone is rapidly absorbed from the upper part of the gastrointestinal tract. Peak serum concentration is reported to occur 45 to 60 minutes following a single dose in fasted patients. This may be extended to 2 hours in fed patients. Following a dose of 25 mg/kg, lower peak serum concentrations have been detected in patients in the fed state (85 μmol/l) than in the fasting state (126 μmol/l), although there was no decrease in the amount of deferiprone absorbed when it was given with food.
Biotransformation :
Deferiprone is metabolised predominantly to a glucuronide conjugate. This metabolite lacks iron-binding capability due to inactivation of the 3-hydroxy group of deferiprone. Peak serum concentrations of the glucuronide occur 2 to 3 hours after administration of deferiprone.
Elimination :
In humans, deferiprone is eliminated mainly via the kidneys; 75 % to 90 % of the ingested dose is reported as being recovered in the urine in the first 24 hours, in the form of free deferiprone, the glucuronide metabolite and the iron-deferiprone complex. A variable amount of elimination via the faeces has been reported. The elimination half-life in most patients is 2 to 3 hours.
INDICATIONS :
DEFERIPRONE CAPSULES are used in treatment of iron overload in patients with thalassaemia major in whom desferrioxamine is contraindicated or is inadequate. Blood dyscrasias, particularly agranulocytosis, have been reported with deferiprone.
Administration :
DEFERIPRONE CAPSULES is for oral administration.
Dosage :
In adults and children the optimum dose of DEFERIPRONE CAPSULES to achieve a negative iron balance is 75 mg/kg/day to be administered in 2-4 divided doses. In some patients a lesser dose of 50 mg/kg/day may be adequate while in others the dose may be increased to 100 mg/kg/day. A total daily dose above 100 mg/kg body weight is not recommended because of the potentially increased risk of adverse reactions. The effect of deferiprone in decreasing the body iron is directly influenced by the dose and the degree of iron overload. After starting deferiprone therapy, it is recommended that serum ferritin concentrations, or other indicators of body iron load, be monitored every two to three months to assess the long-term effectiveness of the chelation regimen in controlling the body iron load. Dose
adjustments should be tailored to the individual patient’s response and therapeutic goals (maintenance or reduction of body iron burden). Interruption of therapy with deferiprone should be considered if serum ferritin measurements fall below 500 μg/l.
CONTRAINDICATION :
• Hypersensitivity to the active substance or to any of the excipients.
• History of recurrent episodes of neutropenia.
• History of agranulocytosis.
• Pregnancy
• Breast-feeding
• Due to the unknown mechanism of deferiprone-induced neutropenia, patients must not take medicinal products known to be associated with neutropenia or those that can cause agranulocytosis.
WARNINGS AND PRECAUTIONS :
Neutropenia/Agranulocytosis :
Deferiprone has been shown to cause neutropenia, including agranulocytosis. The patient’s neutrophil count should be monitored every 3-4 weeks. Data from clinical trials indicate that monitoring of the neutrophil count has been effective in identifying cases of neutropenia and agranulocytosis. Neutropenia and agranulocytosis resolved once therapy was withdrawn. If the patient develops an infection while on deferiprone, therapy should be interrupted and the neutrophil count monitored more frequently. Patients should be advised to report immediately to their physician any symptoms indicative of infection such as fever, sore throat and flu-like symptoms. Suggested management of cases of neutropenia is outlined below. It is recommended that such a management protocol be in place prior to initiating any patient on deferiprone treatment. Treatment with deferiprone should not be initiated if the patient is neutropenic. The risk of agranulocytosis and neutropenia is higher if the baseline absolute neutrophil count (ANC) is less than 1.5 x 109/l.
In the event of neutropenia :
Instruct the patient to immediately discontinue deferiprone and all other medicinal products with a potential to cause neutropenia. The patient should be advised to limit contact with other individuals in order to reduce the risk of infection. Obtain a complete blood cell
(CBC) count, with a white blood cell (WBC) count, corrected for the presence of nucleated red blood cells, a neutrophil count, and a platelet count immediately upon diagnosing the event and then repeat daily. It is recommended that following recovery from neutropenia, weekly CBC, WBC, neutrophil and platelet counts continue to be obtained for three consecutive weeks, to ensure that the patient recovers fully. Should any evidence of infection develop concurrently with the neutropenia, the appropriate cultures and diagnostic
procedures should be performed and an appropriate therapeutic regimen instituted.
In the event of severe neutropenia or agranulocytosis :
Follow the guidelines above and administer appropriate therapy such as granulocyte colony stimulating factor, beginning the same day that the event is identified; administer daily until the condition resolves. Provide protective isolation and if clinically indicated, admit patient to the hospital. Limited information is available regarding rechallenge. Therefore, in the event of neutropenia, rechallenge is not recommended. In the event of agranulocytosis, rechallenge is contraindicated.
Plasma Zn2+ concentration :
Monitoring of plasma Zn2+ concentration, and supplementation in case of a deficiency, is recommended.
HIV positive or other immune compromised patients :
No data are available on the use of deferiprone in HIV positive or in other immune compromised patients. Given that deferiprone can be associated with neutropenia and agranulocytosis, therapy in immune compromised patients should not be initiated unless potential benefits outweigh potential risks.
Renal or hepatic impairment and liver fibrosis :
There are no data available on the use of deferiprone in patients with renal or hepatic impairment. Since deferiprone is eliminated mainly via the kidneys, there may be an increased risk of complications in patients with impaired renal function. Similarly, as deferiprone is
metabolised in the liver, caution must be exercised in patients with hepatic dysfunction. Renal and hepatic function should be monitored in this patient population during deferiprone therapy. If there is a persistent increase in serum alanine aminotransferase (ALT), interruption of deferiprone therapy should be considered. In thalassaemia patients there is an association between liver fibrosis and iron overload and/or hepatitis C. Special care must be taken to ensure that iron chelation in patients with hepatitis C is optimal. In these patients careful monitoring of liver histology is recommended.
Discoloration of urine :
Patients should be informed that their urine may show a reddish/brown discoloration due to the excretion of the iron-deferiprone complex.
Chronic overdose and neurological disorders :
Neurological disorders have been observed in children treated with 2.5 to 3 times the recommended dose for several years. Prescribers are reminded that the use of doses above 100 mg/kg/day are not recommended.
Pregnancy :
Deferiprone is not recommended for use in pregnant women. Women of childbearing potential should be advised to avoid pregnancy due to the mutagenic and clastogenic properties of the product. These women should be counseled to take contraceptive measures and should be advised to immediately stop taking deferiprone should they become pregnant or plan to become pregnant.
Nursing mothers :
It is not known whether deferiprone is secreted into breast milk. In such cases, benefits to the mother must be weighed against the risks to the child.
Paediatric use :
There is no clinical experience of DEFERIPRONE CAPSULES in children below two years of age. Therefore its use is not recommended.
INTERACTIONS :
Due to the unknown mechanism of deferiprone-induced neutropenia, patients must not take medicinal products known to be associated with neutropenia or those that can cause agranulocytosis. Interactions between deferiprone and other medicinal products have not been reported. However, since deferiprone binds to metallic cations, the potential exists for interactions between deferiprone and trivalent cation-dependent medicinal products such as aluminium-based antacids. Therefore, it is not recommended to concomitantly ingest aluminium-based antacids and deferiprone. The safety of concurrent use of deferiprone and vitamin C has not been formally studied. Based on the reported adverse interaction that can occur between deferoxamine and vitamin C, caution should be used when administering deferiprone and vitamin C concurrently.
SIDE EFFECTS :
Agranulocytosis and Neutropenia :
The most serious side effect of therapy reported in clinical trials with deferiprone is agranulocytosis (neutrophils < 0.5 X 109/l) with an incidence of 1.2 % (0.6 cases per 100 patient years of treatment). The observed incidence of the less severe form of neutropenia
(neutrophils < 1.5X109/l) is 6.5 % (3.5 cases per 100 patient years). This rate should be considered in context of the underlying elevated incidence of neutropenia in thalassaemia patients, particularly in those with hypersplenism.
Discolouration Of Urine :
The most common side effect reported with deferiprone was reddish brown urine, reported to be due to the excretion of the iron-deferiprone complex.
Gastrointestinal Tract :
Other common effects include : nausea, vomiting, abdominal pain and increased appetite. These effects are more frequent at the beginning of therapy with deferiprone and in most patients are resolved within a few weeks without the discontinuation of treatment. In some patients it may be beneficial to reduce the dose of deferiprone and then scale it back up to the total 75 mg/kg/ day. Episodes of diarrhoea, mostly mild and transient, have been reported in patients treated with deferiprone.
Arthropathy :
Joint pains have been reported in 10-30 % of patients receiving deferiprone. These usually involve knees, ankles, elbows, hip and the lower back. In a few patients the small joints of the hands and feet have been involved. In some patients swelling of the joints with effusion has been seen. The exact mechanism of this arthropathy is not known, but it appears to be by a mechanism other than immunological. Joint pains have been seen more commonly in patients with a higher serum ferritin and those on higher dose of deferiprone. These may be related to deposition of iron in the joints (due to the chelation process) leading to an inflammatory reaction in the joints. If joint pains occur, it may be necessary to stop the drug for a short while or reduce the dosage. The joint pains usually disappear and the
drug may be restarted at lower doses. Concomitant use of nonsteroidal anti-inflammatory drugs (NSAIDs) such as ibrupofen or diclofenac may be used to control the pain. In a few patients however, joint pains may recur on restarting the drug and thus such patients may be unable to tolerate the drug.
Elevated Liver Enzymes :
Increased ALT values have been reported in patients taking deferiprone. In the majority of these patients this increase was asymptomatic and transient, and their ALT values returned to baseline without discontinuation or decreasing the dose of deferiprone.
Zinc Depletion :
Zinc depletion has been reported on prolonged treatment leading to dermatopathy in 1 % of patients. This can be easily corrected by giving zinc supplements.
Patient Monitoring :
The minimum monitoring which is essential for deferiprone
therapy is as follows :
• Haemoglobin, total and differential white cell counts and platelet counts at 1-2 weekly intervals or as recommended by the treating physician.
• Serum ferritin at 3-4 monthly intervals or as recommended by the treating physician.
OVERDOSAGE :
No cases of acute overdose have been reported. However, neurological disorders (such as cerebellar symptoms, diplopia, lateral nystagmus, psychomotor slowdown, hand movements and axial hypotonia) have been observed in children who had been voluntarily prescribed more than 2.5 times the maximum recommended dose of 100 mg/kg/day for several years. The neurological disorders progressively regressed after deferiprone discontinuation. In case of overdose, close clinical supervision of the patient is required.
STORAGE :
Store below 30°C ( 86°F), protected from moisture and light.
Do not refrigerate.
SHELF LIFE :
24 months from the date of manufacture.
PRESENTATION :
DEFERIPRONE CAPSULES contain deferiprone 250 mg.
3 strips of 10 Capsules per Box.
DEFERIPRONE CAPSULES contain deferiprone 500 mg.
10 strips of 10 Capsules per Box.
Disclaimer : For the use of a Registered Medical Practitioner or a Hospital or a Institution only. Also it is not intended to be used by healthcare professionals or patients for the purpose of prescribing or administering these products. Questions regarding the complete and current content of product labeling / specification / presentation should be directed to SGPharma.